
"Dimensionality reduction is key to solving many real-world problems." - Dr. Fei-Fei Li
Overview
Dimensionality reduction is a crucial tool in the field of computational biology, allowing researchers to efficiently analyze large datasets and identify meaningful patterns in complex biological systems. With the advent of high-throughput technologies, such as next-generation sequencing and microarrays, the amount of biological data being generated has increased dramatically in recent years, leading to an urgent need for effective methods to analyze and visualize this information.
Dimensionality reduction techniques are designed to reduce the number of variables in a dataset, while retaining as much of the relevant information as possible. In computational biology, these techniques are used to reduce the dimensionality of gene expression data, protein-protein interaction networks, and other large-scale biological datasets. By reducing the dimensionality of these datasets, researchers can more easily identify patterns, clusters, and other important features, without being overwhelmed by the sheer amount of data. Additionally, reducing the dimension shortens the time needed for algorithms to run on the data.
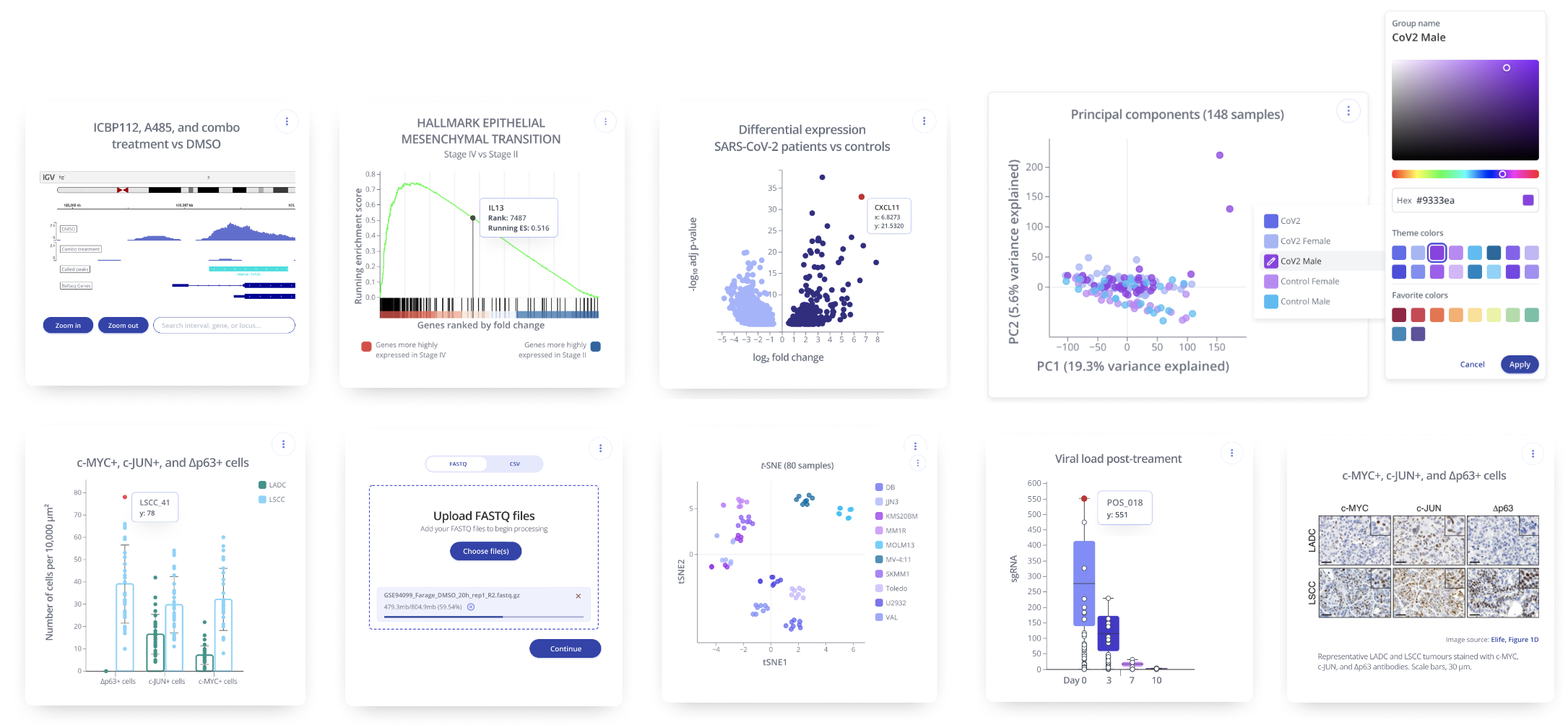
One of the most commonly used dimensionality reduction techniques in computational biology is principal component analysis (PCA). PCA is a mathematical method that transforms a dataset into a set of new variables, known as principal components, which are linear combinations of the original variables. These principal components are ordered in terms of their variance, with the first component explaining the most variance in the data, followed by the second component explaining the most variance when the first component is removed, and so on.
PCA can be used to visualize high-dimensional data in a two- or three-dimensional space, allowing researchers to identify clusters or patterns in the data. It can also be used to identify genes or proteins that are driving the variation in the dataset. For example, PCA can be used to identify the genes that are most strongly associated with a particular disease or phenotype.
Learn more about creating a PCA plot in Pluto.
Another commonly used dimensionality reduction technique in computational biology is t-SNE (t-Distributed Stochastic Neighbor Embedding). t-SNE is a nonlinear dimensionality reduction technique that is particularly effective at visualizing high-dimensional data in a two- or three-dimensional space. It is often used to visualize gene expression data, protein-protein interaction networks, and other types of biological data.
t-SNE works by constructing a probability distribution over pairs of high-dimensional points in such a way that similar points are assigned a higher probability of being chosen than dissimilar points. It then constructs a similar probability distribution over the corresponding points in a lower-dimensional space, and minimizes the Kullback-Leibler divergence between the two distributions. t-SNE is generally used to determine whether the data is separable into clusters by distribution as a preliminary step.
Other dimensionality reduction techniques that are commonly used in computational biology include non-negative matrix factorization (NMF), multidimensional scaling (MDS), and independent component analysis (ICA). Each of these techniques has its own strengths and weaknesses, and the choice of technique will depend on the specific goals of the analysis.
Origin
The origins of dimensionality reduction in computational biology can be traced back to the development of principal component analysis (PCA) in the early 20th century. PCA was initially developed by Karl Pearson in 1901 as a statistical method for analyzing data with many variables. However, it wasn't until the 1960s and 1970s that PCA began to be applied to biological data, such as gene expression data.
In the early days of gene expression microarrays, it became clear that these platforms produced vast amounts of data that were difficult to analyze using traditional statistical methods. One of the key challenges was that the number of genes measured by the microarray far exceeded the number of samples, leading to the so-called "curse of dimensionality". This is because each gene is a feature, increasing the complexity of analysis as they are considered individually.
To address this problem, researchers began to explore the use of PCA as a method for reducing the dimensionality of gene expression data. By applying PCA to the data, they were able to identify patterns of gene expression that were associated with different biological processes or phenotypes.
Over time, other dimensionality reduction techniques, such as multidimensional scaling (MDS), independent component analysis (ICA), and non-negative matrix factorization (NMF), were also developed and applied to biological data. These techniques allowed researchers to identify more complex patterns in the data, such as nonlinear relationships and higher-order interactions.
Today, dimensionality reduction is an essential tool in computational biology, used in a wide range of applications, including gene expression analysis, protein-protein interaction networks, and single-cell RNA sequencing. As the field continues to generate increasingly large and complex datasets, the importance of dimensionality reduction techniques is only likely to grow.
Future of dimensionality reduction
The future of dimensionality reduction in computational biology is likely to be shaped by several key trends and developments. These include:
-
Advances in machine learning: Machine learning algorithms, such as deep learning and neural networks, are becoming increasingly sophisticated and powerful. These algorithms are well-suited to analyzing large-scale biological datasets and can often outperform traditional statistical methods. As machine learning techniques continue to advance, it is likely that they will become an increasingly important tool for dimensionality reduction in computational biology.
-
Integration of different data types: Computational biologists are increasingly working with datasets that contain multiple types of data, such as genomic, transcriptomic, and proteomic data. To analyze these complex datasets, dimensionality reduction techniques will need to be developed that can effectively integrate and visualize different types of data. These techniques should preserve information while being efficient.
-
Single-cell data analysis: Single-cell RNA sequencing (scRNA-seq) is a rapidly growing field in computational biology. ScRNA-seq datasets can contain millions of cells, each with thousands of gene expression values. To analyze these large and complex datasets, new dimensionality reduction techniques will be needed that can accurately capture the heterogeneity and complexity of single-cell data.
-
Privacy concerns: As biological data becomes more widely available, privacy concerns will become increasingly important. Dimensionality reduction techniques will need to be developed that can effectively reduce the dimensionality of datasets while maintaining the privacy of individual subjects.
Overall, the future of dimensionality reduction in computational biology is likely to be shaped by continued advances in machine learning, the integration of different data types, the growth of single-cell data analysis, and increasing concerns around privacy. As these trends continue to evolve, new dimensionality reduction techniques will need to be developed that can effectively analyze and visualize large-scale biological datasets.
Insights via dimensionality reduction
Dimensionality reduction techniques in computational biology can provide a range of biological insights by identifying patterns, clusters, and other important features in large-scale biological datasets. Some of the biological insights that can be gained from dimensionality reduction include:
-
Identifying disease subtypes: By clustering samples based on their gene expression profiles, dimensionality reduction techniques can be used to identify distinct subtypes of diseases, each with its own underlying molecular mechanisms and potential therapeutic targets.
-
Identifying gene expression signatures: Dimensionality reduction techniques can be used to identify gene expression patterns that are associated with specific biological processes, such as cell cycle progression, apoptosis, or immune response.
-
Identifying biomarkers: By identifying genes or proteins that are most strongly associated with a particular disease or phenotype, dimensionality reduction techniques can be used to identify potential biomarkers that could be used for early detection or diagnosis.
-
Visualizing complex biological systems: Dimensionality reduction techniques, such as t-SNE, can be used to visualize high-dimensional data in a two- or three-dimensional space, allowing researchers to identify clusters or patterns in the data and gain new insights into the underlying biology of complex biological systems. This can help determine appropriate downstream analysis steps.
-
Integrating different types of biological data: By reducing the dimensionality of multiple types of biological data, such as genomic, transcriptomic, and proteomic data, dimensionality reduction techniques can be used to integrate different data types and identify new relationships between them.
Overall, dimensionality reduction techniques in computational biology can provide a range of important biological insights that can help researchers understand the underlying mechanisms of diseases, identify potential biomarkers, and develop new treatments.
Dimensionality Reduction in Pluto
Within the Pluto platform a wide variety of analysis types are available, including PCA and t-SNE. Learn more here or contact Pluto for a personalized demo.
Conclusion
In conclusion, dimensionality reduction techniques have become essential tools in the field of computational biology, allowing researchers to efficiently analyze large-scale biological datasets and identify meaningful patterns and features. From the early development of principal component analysis to the latest advances in machine learning and single-cell data analysis, dimensionality reduction techniques have played a critical role in advancing our understanding of complex biological systems. As the field of computational biology continues to generate increasingly large and complex datasets, the importance of dimensionality reduction techniques is only likely to grow. By harnessing the power of these techniques, researchers can gain new insights into the underlying biology of diseases and other biological processes, paving the way for new therapies, biomarkers, and diagnostic tools.