Pluto Bioinformatics
GSE124822: DNA methylation disruption reshapes the hematopoietic differentiation landscape
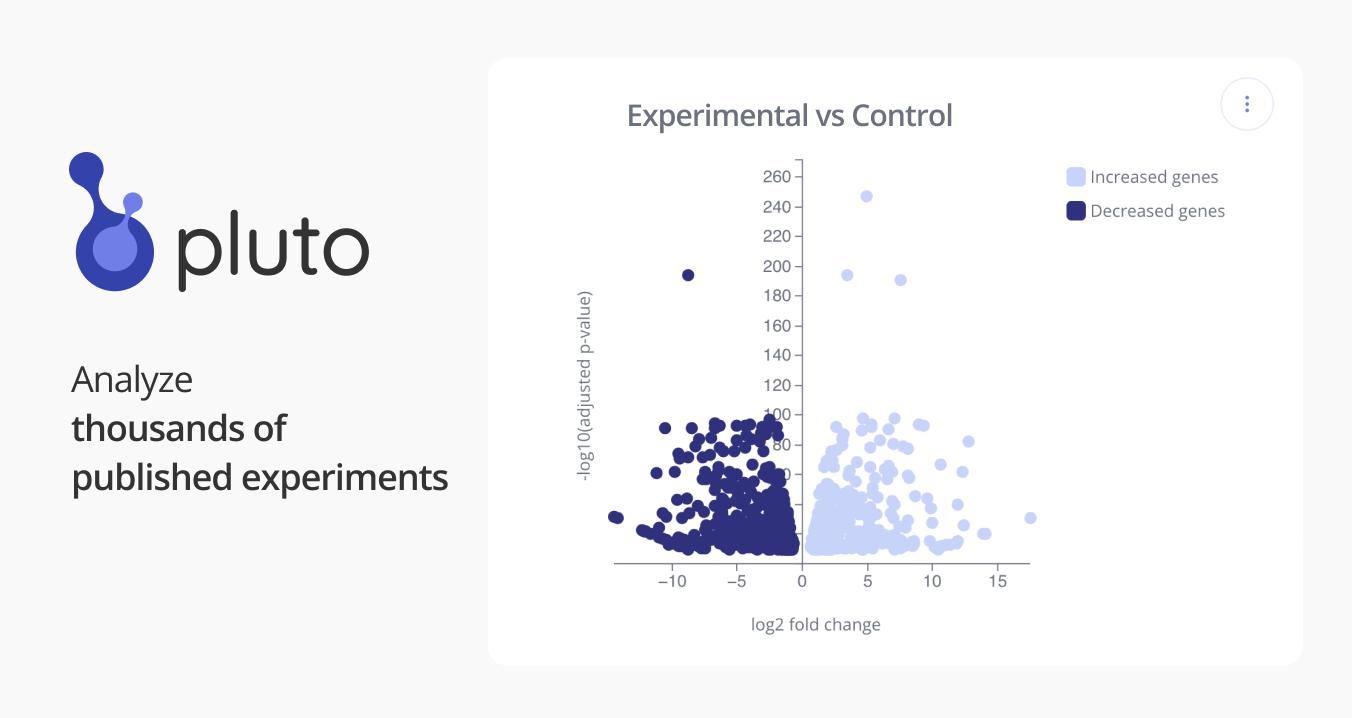
Bulk RNA sequencing
Somatic mutations in genes implicated in DNA methylation (DNAme; e.g., TET2, DNMT3A and IDH2), are frequently observed in hematological malignancies, as well as in clonal hematopoiesis (CH). Yet, how these mutations disrupt the hematopoietic differentiation topology remains largely unknown. By applying complementary single-cell sequencing approaches to murine bone marrow hematopoietic stem and progenitor cells, we observe that mutations in DNAme modifiers result in significantly altered differentiation topology. In particular, we find shifts in the frequencies of erythroid vs. myelo-monocytic progenitors, which can be traced back to fate-priming skews in the earliest uncommitted hematopoietic stem cells (HSCs). DNAme analysis both in open chromatin regions and at single-cell resolution, demonstrate that methylation changes resulting from DNAme modifier disruption are distributed stochastically across the genome. To reconcile the distributed nature of DNAme changes with the deterministic erythroid vs. myelo-monocytic topological skews, our data implicate differential sensitivity of key transcription factors to global methylation changes due to biases in CpG enrichment in their binding motifs. Finally, by coupling single-cell whole transcriptome analysis with targeted genotyping, we observe a similar skew in early transcriptional priming impact human CH bone marrow progenitors in an individual with DNMT3A-755S mutation. Thus, our results provide evidence that DNAme encodes topological information for hematopoietic differentiation. SOURCE: Franco Izzo (fri2002@med.cornell.edu) - Landau Weill Cornell Medicine